Magnetic Structures in GSAS-II – Tutorial I
Introduction
In this and subsequent tutorials, we will show how GSAS-II makes use of the Bilbao crystallographic Server (www.cryst.ehu.es/) Magnetic Symmetry and Applications routine k-SUBGROUPSMAG (Perez-Mato, JM; Gallego, SV; Elcoro, L; Tasci, E and Aroyo, MI J. of Phys.: Condens Matter (2016), 28:28601) to determine magnetic crystal structures. If you have not done so already, you may wish to do the Simple Magnetic Structures tutorial first as it contains some background information that will not be covered here. To make this work, your computer must have an internet connection for GSAS-II to access this site. (We will supply the project file that results after k-SUBGROUPSMAG is called in case you lack internet access)
Note that to access the Bilboa Crystallographic Server from inside GSAS-II you must register for access. This only needs to be done once. See the Setup Access to the Bilbao Crystallographic Server tutorial for details on how to do this.
Starting with the grey group for the parent chemical structure (e.g. Pnma1’) and any propagation vector (k-vector), k-SUBGROUPSMAG operates by removing symmetry operators to generate magnetic space subgroups. The remaining operators in each case are identified with a standard setting of the known magnetic BNS space groups (2nd setting with inversion at the origin as needed). It generally doesn’t stop until the lowest possible symmetry (triclinic) is reached; there can be more than 100 of these possible subgroups. For each one, the magnetic space group symbol, lattice transformation matrix and any required translation of the original coordinates is returned in a table of the results. In GSAS-II, a special version of the k-SUBGROUPSMAG web page is called with the parent chemical space group operations, your selected k-vectors and some option flags. It return an html file containing a table which is parsed by GSAS-II to extract the generated magnetic space groups, transformation matrices, origin shift vector, conjugacy classes (if any) and super group lists. If the resulting space group is orthorhombic, GSAS-II will by default transform the standard setting to a setting where the unit cell axes orientation is preserved vs the chemical cell. GSAS-II will also optionally examine the magnetic atom positions to determine if any or all can carry a nonzero moment, flagging those space groups with atoms that can. This simplifies the search for a correct description of the magnetic structure.
To see how this works for a simple case, we will repeat the first exercise in the Simple Magnetic Structures in GSAS-II tutorial in this tutorial, skipping some steps as these are not needed. We will redetermine the magnetic structure for the simple antiferromagnet LaMnO3 which is a perovskite distorted by a strong Jahn-Teller effect to an orthorhombic cell. Neutron powder diffraction data was obtained on BT-1 at the NIST reactor at 50K (thanks to Qingzhen Huang for providing the data).
If you have not done so already, start GSAS-II (make sure about the internet connection!).
Simple Antiferromagnet: LaMnO3
Step 1: Read in the data file
1. Use
the Import/Powder Data/from GSAS powder data file menu item
to read the data file into the current GSAS-II project. This read option is set
to read any of the powder data formats defined for GSAS (angles in centidegrees, TOF in µsec). Other submenu items will read
the cif
format or the xye
format (angles in degrees) used by Topas, etc. In those cases, you would change the file
directory to cif
format or the xye
format (angles in degrees) used by Topas, etc. to see
them. Because you used the Help/Download tutorial menu entry to open this page and
downloaded the exercise files (recommended), then the Magnetic-I/data/...
entry will bring you to the location where the files have been downloaded. (It
is also possible to download them manually from https://advancedphotonsource.github.io/GSAS-II-tutorials/Magnetic-I/data/.
In this case you will need to navigate to the download location manually.)
For this tutorial you should see the data file in the file browser, but if
extensions on data files are not the expected ones, you may need to change the
file type to All files (*.*) to
find the desired file.
2. Select the LaMnO3_50K.gsas data file in the first dialog and press Open. There will be a Dialog box asking Is this the file you want? Press Yes button to proceed.
3. Since this data file does not define an instrument parameter file, a file dialog will appear for you to select the correct instrument parameter file. Select BT1_Cu311.inst; you may have to change the file type in the file selection Dialog box to GSAS iparm file to see it. At this point the GSAS-II data tree window will have several entries
and the plot window will show the powder pattern
Step 2: Select Limits
This pattern has more data than we need, so it is helpful to cut down the range and one probably doesn’t want to use the two very broad peaks at the upper end of the pattern as they don’t contain much information for the magnetic structure. One also should be careful in selecting the lower limit especially for magnetic structure studies as a small peak may be hidden at low angles that can decisively determine a magnetic structure (there are none in this example, but this issue will be apparent in the next example). Click on the Limits item in the GSAS-II data tree. Use the New: set of entry boxes to set Tmin and Tmax to 6 and 156. Notice on the plot that the limit lines have moved to these positions; the green lower limit is just below the 1st peak and the red upper limit is just below that last two broad peaks. You may also drag the limit lines to the desired location or place them by a left mouse click on a data point for the lower limit or a right click on a data point for the upper limit. The plot should look like
Step 3: Read in the chemical structure for LaMnO3
1. Use the Import/Phase/from CIF file menu item to read the phase information for LaMnO3 into the current GSAS-II project. This read option is set to read Crystallographic Information Files (CIF). Other submenu items will read phase information in other formats. Because you used the Help/Download tutorial menu entry to open this page and downloaded the exercise files (recommended), then the Magnetic-I/data/... entry will bring you to the location where the files have been downloaded. (It is also possible to download them manually from https://advancedphotonsource.github.io/GSAS-II-tutorials/Magnetic-I/data/. In this case you will need to navigate to the download location manually.)
2. Select the LaMnO3.cif data file in the first dialog and press Open. There will be a Dialog box asking Is this the file you want? Press Yes button to proceed. You will get the opportunity to change the phase name next (I took out the spaces!); press OK to continue.
3. Next is the histogram selection window; this connects the phase to the data so it can be used in subsequent calculations.

Select
the histogram (or press Set All)
and press OK. The General tab for the phase is shown next

Step 4. Check powder pattern indexing
Here the objective is to determine how the reflection positions generated from the chemical (“nuclear”) crystal structure lattice match up with the peaks observed for this antiferromagnet.
1. Select
Unit Cells List from the tree entries under the
histogram (begins with PWDR); the Indexing controls will be shown
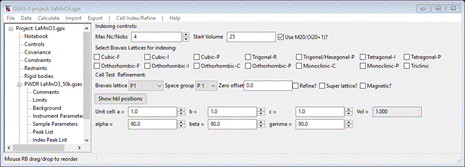
This can be use lattice parameters & space groups to generate expected reflection positions to check against the peaks in the powder pattern.
2. One
could then enter Bravais lattice, choose space group
& enter lattice parameters by hand, but the easy way is to use the chemical
lattice directly. Do Cell
Index/Refine/Load Cell from the menu; a Phase selection box will
appear with only one choice (LaMnO3).
This was taken from the Phases present in this project; there could be more
than one depending on what you loaded/created earlier. The Import
menu item allows you to get lattice information from any one of the other phase
containing files known to GSAS-II. Press OK
and the Indexing controls will be change
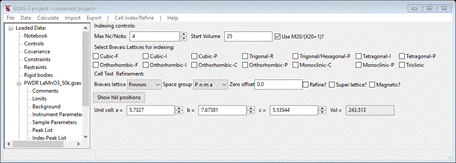
showing the lattice constants for LaMnO3 you had obtained from the cif file and the powder pattern will change showing the
lines for P n m a and the LaMnO3 lattice parameters. Change the
space group to P m m m; the plot will change showing the new lines.

Notice that every peak is indexed by this cell, therefore the magnetic lattice is the same as the chemical cell. It just has some magnetically allowed reflections that were forbidden in the space group P n m a. No doubling of the unit cell along any axes is needed, this makes the magnetic propagation vector, kx ky kz = 0,0,0; this will be needed in the next step. Now change the space group back to P m n a (VERY IMPORTANT!) or else repeat the Cell Index/Refine/Load Cell done in step 2 above to recover the LaMnO3 data.
Step 5. Run k-SUBGROUPSMAG
In this next step, you will be accessing the Bilbao crystallographic server so you must be connected to the internet. In the Cell Index/Refine menu select Run k-SUBGROUPSMAG; a small popup dialog will appear
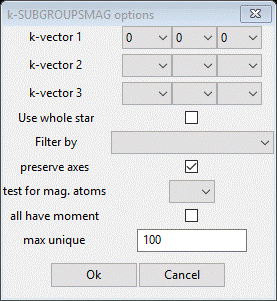
These are the controls for running the Bilbao routine. The space group is taken from the Space Group entry on the Unit Cells window. Since we did not discover any cell doubling, leave the kx ky kz entries as they are. Do select Mn from the test for mag. atoms pulldown as that will restrict the models to those with a nonzero magnetic moment on the Mn atoms. We will discuss the other options in another tutorial. Press Ok. A popup dialog will appear reminding you how to cite the Bilbao routine in any paper you write using this facility.

Press Ok,
the nag note will also appear on the console and after a pause, the console
will show “request OK” indicating that the Bilbao site has responded with some
results. GSAS-II will next display a File Dialog asking for a project name to
save this project; change the directory to a useful place for your work on this
tutorial and give the project a name; I used LaMnO3
base.
The GSAS-II data window will display the results from k-SUBGROUPSMAG. (If k-SUBGROUPSMAG failed because you lack internet access, this file is located in the same location as the powder data and cif files used above were found. Do File/Open Project from the main menu to access it “LaMnO3 base.gpx”).
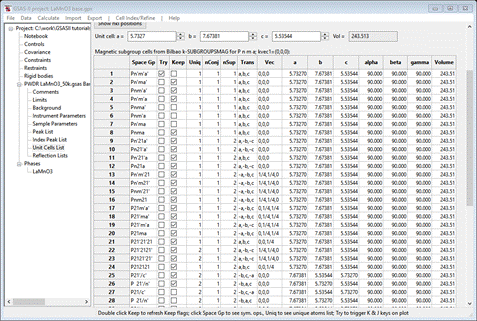
The table shows all the magnetic space groups that are possible starting with P n m a and zero propagation vector; this is noted in the table heading. Notice that it goes all the way to a triclinic P1 magnetic space group; you probably will never need to go that far to find a suitable magnetic space group. The ones marked ‘Keep’ have a possibly magnetic Mn atom based on the magnetic site symmetries of the transformed atom positions (there may be more than one). The Uniq column indicates how many unique Mn atoms are present in the magnetic structure; usually one wants to select one with the smallest number of magnetic sites. Each entry also shows the transformation matrix in symbolic form and the necessary origin shift so that the new structure is properly positioned to satisfy the new space group. nConj indicates how many other magnetic space groups are members of the conjugate class (in this case only 1 for every space group) and nSup shows how many super groups there are for each space group. Those with only one may have just the parent space group as their super group; these are the “maximal” subgroups resulting from the loss of just a single symmetry generator. The orthorhombic ones (1-24) are all transformed so the cell matches the parent; this is the default action. K-SUBGROUPSMAG returned these in the standard orthorhombic setting; GSAS-II reset these to match the parent. The Try box is for you to test the reflection indexing of each structure to be sure all the magnetic peaks are included. Try each of the first 3 that are marked Keep (you may recognize these from the Simple Magnetic tutorial). As you select each one the powder pattern will be replotted showing the allowed lines. For the 2nd one, Pn’ma’, the plot shows all lines indexed (apart from the contaminant peak at 15.69°2Θ).

I’ve zoomed in on the low angle region for clarity. If you Try the Pnma one as well, it will index all the lines (even showing two that straddle the contaminant line). You can then un”Keep” the other 2 choices so they don’t appear in a later step where you select the magnetic phase. Now do a Save project; that will save your choices for later (there should be at least two).
Step 6. Select a magnetic phase
In the previous step we discovered two possible highest symmetry magnetic structures that both indexed all the reflections in the powder pattern (in one case even the contaminant line) and allowed the Mn atom sites to have magnetic moments. We will see those next. Select the LaMnO3 phase from the GSAS-II data tree (under Phases). The General tab is shown for this nuclear phase.

Do Compute/Select magnetic/subgroup phase; that will make a small popup dialog
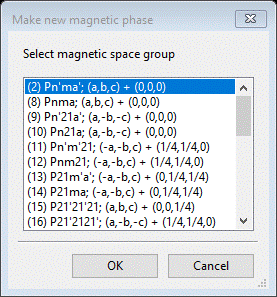
This shows all magnetic phases that were marked Keep in the Unit Cells table; for each you see the transformation and vector (occasionally two results will have the same space group but have different vectors). Select the 1st one and press OK; a new popup dialog will appear.

This shows the magnetic atoms in this structure and allows one to reject certain ones that are known to not be magnetic. If you press Delete then this space group will be unmarked Keep in the Unit Cell List and will not be available for a future selection. If you select No then nothing will happen and you can find this space group in a subsequent try of Select magnetic phase. This space group is the desired one; press Yes to select it. The project will be saved first, then a File Dialog box will appear offering a new project file name (LaMnO3 mag_2.gpx) for saving the project and renaming it. It will be in the same directory as the original one (LaMnO3 base.gpx). Press Save; the new project will be opened to the General tab for magnetic phase just created.

The phase is named with “mag_2” appended to the end, the phase type is magnetic and there are magnetic spin operator selections part way down the page. These are properly set for the chosen magnetic space group Pn’ma’ as “red”, ”black”, ”red”. You also can see the Lande’ g factor can be changed if needed and it only contains the Mn atom. Notice that the base project is unchanged (and closed) in this process. That allows you to go back to it to try another magnetic phase if necessary (in this case the Pnma phase). Also note that the k-SUBGROUPSMAG results table has been deleted from the Unit Cells List; this keeps you from inadvertently making a second magnetic phase in this project that would clash with the first one. Now, let’s explore the Pn’ma’ magnetic structure.
Step 7. Solving the magnetic structure
1. Test the Pn’ma’ model
So now we have a spin arrangement to consider, Pn’ma’, but we do not know the magnetic moment components, Mx, My & Mz. However, since the 1st reflection (the 010) is magnetic and strong, it is likely that the value of My=0. We can force this (to avoid refinement problems) by making a ‘hold’ on its value. Select the Constraints item from the GSAS-II tree; it will show the constraints generated when the magnetic phase was made

Under the Edit Costr. menu item do Add hold; a Hold phase variable window will appear
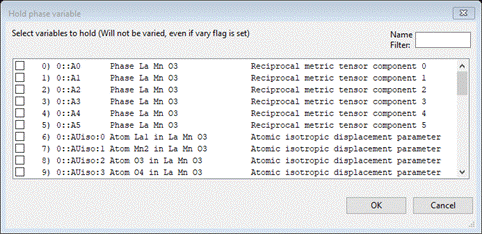
This can be simplified by putting My in the Name Filter box

Select the entry and press OK; the constraint window will show the Hold at the bottom of the list.
Now return to phases and select La Mn O3 mag_2 and the Atoms tab; you should see

The least squares will not begin a refinement of Mx & Mz starting from zero since either +/- values are equally possible; the result will be a singularity. So these values must be offset so set them to 1.0 each. Do Calculate/Refine; the refinement will quickly converge to Rwp ~14% and the powder plot will look like
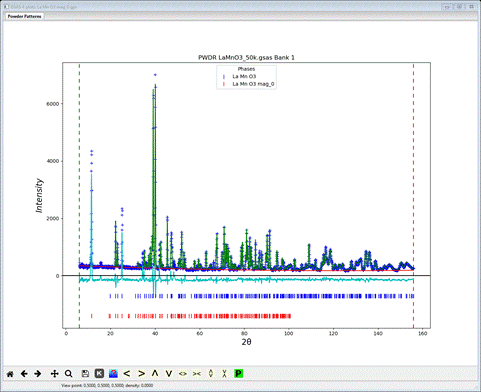
There does not appear to be any calculated magnetic intensity, but a zoom onto the low angle (10-30°2Θ) part of the pattern shows that some intensity was obtained, but clearly not enough.
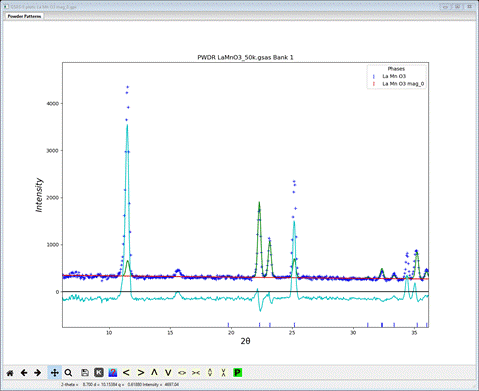
The magnetic moment is clearly too small; to let the least squares find a better value set the refine flag for Mn to ‘M’ and repeat Calculate/Refine. The residual will drop considerably to Rwp ~8.7% and the low angle portion will show a much better fit.
This appears to be a reasonable solution; a more complete refinement still needs to be done, we’ll do that in the next step after we test the Pnma model. Save the project.
2. Test the Pnma model
Now reopen the original LaMnO3 base.gpx file saved at Step 5 above. Select La Mn O3 phase from the GSAS-II tree; the General tab will be shown

Do Compute/Select magnetic phase; the magnetic phase selection box will show again
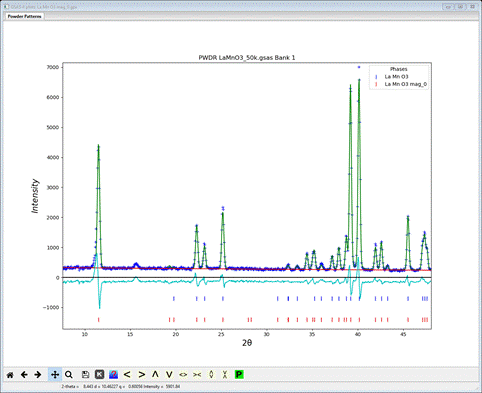
This time select the second choice “(8) Pnma; (a,b,c) + (0,0,0)” and press OK. A new popup dialog will appear
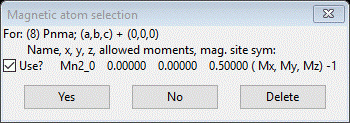
Press Yes to select this choice; a File dialog will appear with a new project name for this magnet model (LaMnO3 mag_8.gpx). Press Save to select it; the projects will be exchanged opening the new magnetic one showing the Pnma magnetic phase
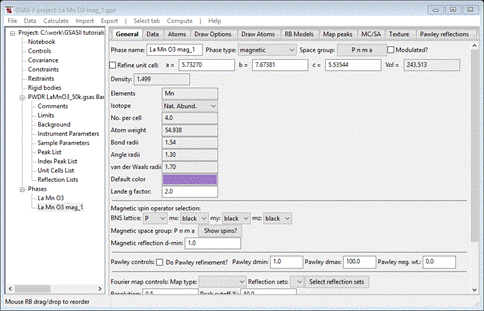
This time all the spin operators are “black”. Select the Atoms tab
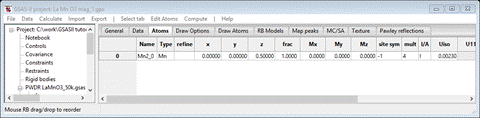
Again, one can make the same argument that since the 010 reflection is large then My should be zero. Set Mx & Mz to 1.0 and then go to Constraints and set the hold on 1::AMy:0. Do Calculate/Refine from the main menu to do a Rietveld refinement. It converges to Rwp ~14%; about the same as the Pn’ma’ model at the same step. The low angle portion of the powder pattern shows

There is intensity in the lowest angle reflection (the 010) but not enough. Again we’ll try to refine the Mx & Mz components to see what happens. Set the refine flag for Mn to ‘M’ and repeat Calculate/Refine. The residual will drop to Rwp ~10% and the low angle portion will show a much better fit which is very similar, but slightly poorer than the result from the Pn’ma’ model. To see how, examine a slightly higher angle region (30-55°2Θ) and compare it with the same region for the plot for the Pn’ma’ model (you’ll need2 instances of GSAS-II running to do this)
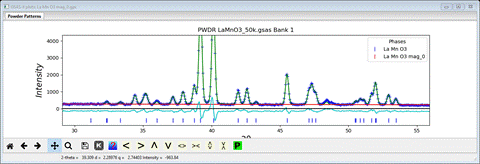

You can see in particular at the pair of peaks at 33.3 and 34.4°2Θ that are well fit by the Pn’ma’ model and not by the Pnma one. On this basis the Pnma model is rejected and the Pn’ma’ is selected as probably correct. NB: neither model produced any intensity for the peak at ~15.69°2Θ, so it is most likely to be from a minor contaminating phase; we can either ignore it or exclude it in the final refinements.
Step 8. Complete refinement of the Pn’ma’ model for LaMnO3
Reopen the La Mn O3 mag_2.gpx project file which should have the Pn’ma’ model for the magnetic structure, and look at the powder pattern to decide how to proceed to complete the refinement.
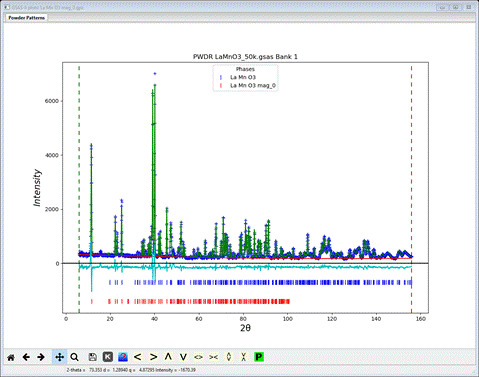
Looking at the plot, it would seem that the lattice parameters and sample position need to be refined. It is important to remember that if a parameter in one phase is varied then the corresponding one in the other phase must also be varied, otherwise GSAS-II will report the error and refuse to attempt the refinement. To set refinement of the lattice parameters, bring up the General tab for each phase & check the Refine unit cell box for the lattice parameters. Then select Sample Parameters for the PWDR entry
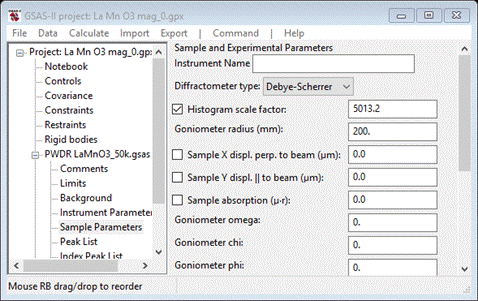
Since the scan covers a very wide range in 2Q, both sample displacements can be refined. Check both Sample X and Y displ parameters. NB: if the correct Goniometer radius (650mm) is used, these displacements will be in microns. Also, it would appear that the instrument parameters do not adequately describe the anisotropic broadening of the lowest angle reflection so we will need to include that. Select Instrument Parameters from the PWDR entry and check the U, V, W & SH/L Refine boxes. Now do Calculate/Refine until convergence is achieved (i.e. Rwp doesn’t change). This will be 2-3 times. When done my Rwp was ~6.2% with a nice clean plot

Now we can add refinement of the atom positions and thermal parameters. For Atoms tab of the LaMnO3 phase, double click the refine column heading and select X & U. This will add these parameters as allowed by symmetry to the refinement. Now go to the Atoms tab for La Mn O3 mag_2 and double click the refine tab. Select U & M; the Uiso for the Mn atom is tied via a constraint to the Mn in the other phase. Now select Background from the PWDR entry and increase the number of terms to 6. Finally do Calculate/Refine until convergence (1-2 times). My Rwp was 5.95% with a plot that is hardly different from the previous one. If you press the ‘W’ key with the focus on the plot, GSAS-II will show the weighted difference curve below the plot

This curve shows a couple of peaks which are from some contaminating phase, but otherwise the fluctuations are mostly within 2s of zero. Finally examine the magnetic moment components of the Mn atom; Mz is close to zero. It is likely that it is zero; set it to zero and then go to the Constraints tree item and add a hold for the 1::AMz:0 parameter so that it stays at zero. Repeat the refinement; the residual is unchanged showing that setting Mz=0 was a valid assumption.
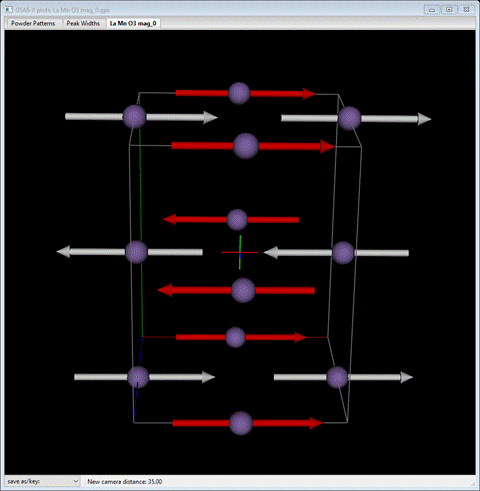
Step 9. Draw the magnetic structure of LaMnO3
To draw the magnetic structure select the Draw Atoms tab for the magnetic phase. The table will have a single line matching that of the Atoms table with a few extra columns.
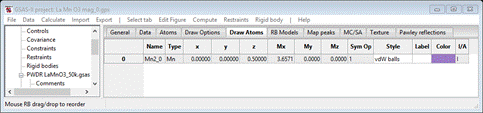
The plot will show one atom with magnetic moment. Select the atom in the table and do Edit Figure/Fill unit cell. The table will show additional entries

The plot will show the unit cell contents of Mn atoms each with its magnetic moment; they are arranged in layers along the b-axis with the moments antiparallel from one layer to the next. Some are red indicating that a spin inversion operation was applied for that atom moment.
This completes this tutorial; you can save the project if you wish & close GSAS-II.
If desired you can try to repeat the 2nd example, Ca doped LaMnO3, which is a ferromagnet. There is no cell doubling so again the propagation vector kx,ky,kz is zero and k-SUBGROUPSMAG will suggest 4 possible magnetic space groups for Pbnm (a nonstandard variant of Pnma) which have an even number of spin inversions. Again, if k-SUBGROUPSMAG is unavailable, you can find the result in LaCaMnO3 base.gpx. There is no distinction based on reflection extinctions, so all 4 must be tested as shown in the Simple Magnetic Structures tutorial. You should find as before that the Pbn’m’ magnetic structure is the best one.